
In the US, Center For Devices And Radiological Health (CDRH) which is a branch of FDA is the regulatory body that governs the rules and regulations for medical devices. It ensures the safety and smooth regulation of medical devices. Medical devices are divided into different categories and each of them requires different regulations which are overlooked by CDRH of FDA.
What Is A Class I Medical Device?
Class I Medical Devices often called “class I devices by FDA” are the devices that possess minimal risk and are associated with lowest regulatory requirements and controls.
Class I devices manufacturers do not require submitting pre-market notification and nor do they need to fulfill 21 CFR Part 820 requirements which the other two classes of medical devices require to follow.
Regulatory Classification for Class I Medical Devices:
The regulatory bodies are the FDA in the US and EU in Europe classify the medical devices are:
- Class I: Low risk and possess little to no harm to patients
- Class II: Moderate risk to patient
- Class III: High risk to patient
1. FDA Medical Devices Classification
Class I Medical Devices by FDA:
Class I: Low risk and possess little to no harm to patients
FDA class 1 medical devices constitute 47% of the total medical devices.
Class I Medical Device Regulations are:
- Design and manufacturing controls
- Quality system regulation
- Labeling requirements
- Post-market surveillance
Examples of Class 1 medical devices are: Scalpels, Most bandages, Oxygen masks, Bedpans, Latex gloves, Manual stethoscopes, etc.
To check and confirm under which category your medical device falls kindly visit the FDA official page.

2. EU Medical Devices Classification
As per EU medical devices are classified as follows:
- Class I Devices
- Class IIa Devices
- Class IIb Devices
- Class III Devices
Here we will understand about Class I Medical Devices in EU:
Class I Devices in EU:
For manufacturers looking to market their medical devices in the EU, understanding the classification and certification process is crucial. These are low risk everyday use medical devices. Manufacturers require to issue the technical document for such devices.
EU Class I medical devices encompass a broad range of non-invasive products that pose the lowest risk to patients. However, Class I is not a one-size-fits-all category; it includes several subclasses, each with its own characteristics:
- General Class I: These devices are non-sterile and have no measuring function, representing the lowest risk category. Examples include wheelchairs, plasters, and hospital beds. These devices are Self-Certified and Notified Body certification is not required for these devices.
- Class Is (Sterile): Devices placed on the market in a sterile condition fall under this subclass. Examples include personal protection kits. Notified Body certification is partially required.
- Class Im (Measuring): Devices with a measuring function belong to this subclass. Examples include stethoscopes, thermometers, and weighing scales. Notified Body certification is partially required.
- Class Ir (Reusable): This new subclass includes products that can be reprocessed or reused in some way, such as surgical instruments and endoscopes. Notified Body certification is partially required.

What Exemptions Exists For Class I Medical Device?
As class I devices possess low risk, they are exempted from many regulatory requirements such as:
- Premarket notification or PMA application for FDA
- Manufacturing Processes (GMPs) for medical devices
- Activities under 21 CFR Part 820
EU Regulatory Process For Class I Medical Devices:
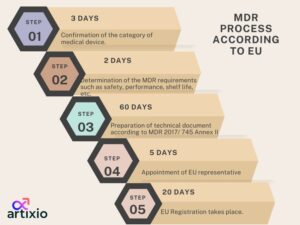
Class I medical device regulations are according to EU that take place in 5 steps beginning from confirmation of medical device category to ending at EU registration. The steps that depict the class 1 medical device requirements are explained in the diagram below:
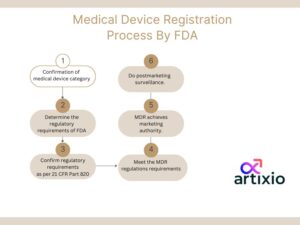
The process according to FDA for MDR begins with the confirmation of medical device category and ending up with marketing approval and post marketing surveillance, which is explained below in the diagram:
Medial Devices CE Certification
The CE marking is a critical standard for medical devices intended for the EU market. It signifies that a device complies with regulatory requirements necessary for entry into European countries. Manufacturers must ensure regulatory compliance and secure CE marking for their products, regardless of whether they outsource any or all components of their manufacturing operations.
CE Certification of Class I Medical Devices
Class 1 devices are generally associated with minimal risk, while medical devices in Classes Is, Im, and Ir are characterised by relatively low to medium levels of perceived risk. The non-sterile and non-measurable Class I devices can obtain CE marking through self-declaration as per EU MDR, eliminating the need for Notified Body certification or approvals from certification bodies. This allows manufacturers to self-certify their devices, making the process more streamlined. Manufacturers of Class I products not requiring Notified Body certification are responsible for creating and maintaining the necessary documentation at their manufacturing facility.


Actions following the placement of a Class 1 Medical Device on the EU market involve gathering and assessing Post Market Surveillance Data, overseeing the Vigilance system, and addressing non-conforming products.
In the event that a manufacturer discovers a device they have released on the market or put into service does not comply with the EU MDR, they will promptly initiate corrective measures to ensure the device’s compliance. This may involve bringing it into conformity, withdrawing it from the market, or initiating a recall, as deemed appropriate.
QMS Requirements for Class I medical device manufacturers
The Class I medical device manufacturer should have established a QMS that includes procedures describing how the following processes are conducted:
- Risk management for medical devices
- Management responsibility and resource management, including the Supplier Management
- Strategy for regulatory compliance, including the conformity assessment process and change management
- Technical documentation management
- Clinical evaluation including Post-market clinical follow-up (PMCF)
- Labeling management
- UDI management
- Product realization
- Post-Market Surveillance (PMS) system and Vigilance reporting Procedures
- Process/product monitoring & measurement and product improvement
- Corrective Actions / Preventive Actions (CAPA)
So, with Artixio you can easily launch your medical device onto the market according to the regulatory requirements of the class in which your medical device fits well. So, contact at info@artixio.com for achieving a successful medical device.
FAQ’s:
Is FDA approval required for Class 1 devices?
No, Class 1 medical devices do not require FDA approval as they are low risk devices.
Do Class I medical devices require premarket approval?
No, Class I medical devices do not require premarket approval.
Can medical devices of class I be sold in EU market?
Yes, medical devices of all classes can be sold in EU markets that comply with EU regulations.



